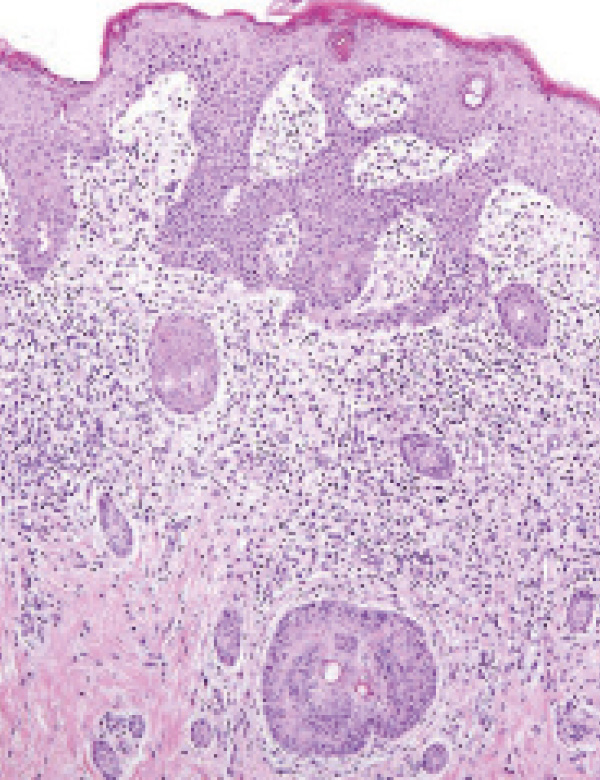
Histiocitosis de células de Langerhans: una actualización basada en un caso real. Parte 2
DOI:
https://doi.org/10.29176/30284163.1961Palabras clave:
Histiocitosis de células de Langerhans, Histiocitosis de células no Langerhans, Reticu lohistiocitosis autoinvolutiva congénita (enfermedad de Hashimoto-Pritzker)Resumen
Las histiocitosis son trastornos raros caracterizados por la acumulación de células dendríticas o macrófagos en diversos órganos, con predilección por piel y hueso (1). Su comportamiento clínico varía desde formas leves hasta potencialmente mortales (2). La primera clasificación de histiocitosis de 1987 (3) constaba de 3 categorías: Células de Langerhans (HCL), no relacionadas con LC (HCNL) e histiocitosis malignas (HM). Estas neoplasias inflamatorias pueden presentarse en formas limitadas, difusas o con compromiso multiorgánico (4), manifestándose de múltiples maneras e imitando a otras enfermedades, por lo que representan un desafío diagnóstico para la clínica.
Biografía del autor/a
Mario Zavala-Mena, Universidad de Santiago de Chile.
Residente de Dermatología, Universidad de Santiago de Chile.
Isabel Jimeno-Ortega, Universidad de Santiago de Chile.
Residente de Dermatología, Universidad de Santiago de Chile.
Gabriel Aedo-Inostroza, Universidad de Santiago de Chile.
Dermatólogo, Universidad de Santiago de Chile.
Cómo citar
Descargas
Publicado
Cómo citar
Número
Sección
Licencia
Derechos de autor 2024 Mario Zavala-Mena, Isabel Jimeno-Ortega, Gabriel Aedo-Inostroza

Esta obra está bajo una licencia internacional Creative Commons Atribución-NoComercial-CompartirIgual 4.0.